C1.0. Overview
The “Crystal Information” component (control) provides functions to set various information about crystals. This control is a common control used in ReciPro, PDIndexer, CSManager, and other software, and does not operate as a standalone module. Please note that in software such as ReciPro, PDIndexer, and CSManager, multiple crystals are used in list form. When a new crystal is created or modified, it will not be reflected in the crystal list until you press the Add button or Replace button in each software. Please note this.
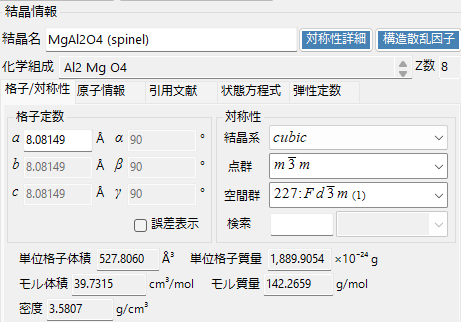
Crystal Name
Set/display the crystal name.
Chemical Composition
The chemical composition of the crystal is displayed. (Only when atomic information is entered)
Z Number
The number of formula units contained in the unit cell is displayed. (Only when atomic information is entered)
C1.1. Lattice/Symmetry Tab
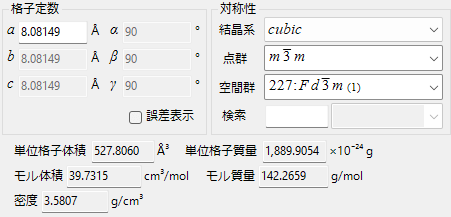
On this tab, you can set/display the lattice constants and symmetry of the crystal.
Lattice Constants
Display/set the lattice constants. The unit is Å (10-10 m). When you specify the symmetry, if there are restrictions on the lattice constants (for example, a=b=c, α=β=γ=90°, etc.), they are automatically reconfigured.
Symmetry
Display/set symmetry at each hierarchical level.
Crystal System
Display/set the crystal system.
Point Group
Display/set the point group.
Space Group
Display/set the space group. Please note axis settings and similar parameters.
Search
Type the space group string and its candidates will appear in the list on the right. Case sensitivity is applied.
Volume, Mass, and Density
Unit Cell Volume
The volume of the unit cell is displayed.
Unit Cell Mass
The weight of the unit cell is displayed.
Molar Volume
The volume per mole is displayed. (Only when atomic information is entered)
Molar Mass
The weight per mole is displayed. (Only when atomic information is entered)
Density
The density is displayed. (Only when atomic information is entered)
Color of Profile (PDIndexer only)
Set the color for drawing diffraction peaks. Clicking opens a window for color settings.
C1.2. Atomic Information Tab
This tab displays and sets the information of atoms contained in the crystal. The upper list shows a list of atoms contained in the crystal, and when you click on each atom, detailed atomic information is displayed in the lower part of the screen. Please note that atomic information is not saved in the list until you press the “Add” or “Update” button.

Add
Add the configured atom to the list as a new entry.
Update
Replace the configured atom with the currently selected atom.
Move Up/Move Down
Move the order of the selected atom up/down.
Delete
Delete the selected atom from the list.
Element Type and Atomic Position
Set the element type and atomic position in the unit cell (fractional coordinates).
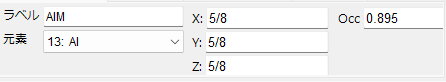
Label
Enter the atom label.
Element
Display/set the element.
X, Y, Z
Display/set the atomic fractional coordinates. Enter real numbers from 0 to 1. You can also enter fractions like 1/2 or 2/3.
Occ
Specify the occupancy of the atom. Specify as a real number from 0 to 1.
Origin Shift
Perform a shift of atomic positions.

Preset Buttons (1/8 1/8 1/8, etc.)
Shift the origin position using preset values. You can change the sign by checking “+” or “-“.
Apply custom shift
Shift the origin position using custom values.
Debye-Waller Factor
Set the Debye-Waller factor (temperature factor).
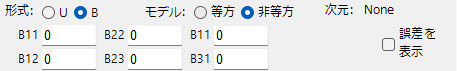
Format
Select either U or B.
Model
Select either isotropic or anisotropic.
B## or U##
Enter the temperature factor.
Scattering Factor
Configure the valence and isotopic composition settings for calculating atomic scattering factors.
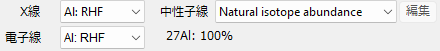
X-ray
Select the atomic valence when calculating the elastic atomic scattering factor for X-rays. Parameters are cited from International Tables for Crystallography volume C.
Electron
Select the atomic valence when calculating the elastic atomic scattering factor for electrons. Parameters are cited from Peng (1998, Acta Cryst A54, 481-485).
Neutron
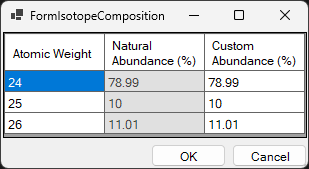
Select the isotopic composition when calculating the elastic scattering length of neutrons. You can select either “Natural isotope abundance” or “Custom isotope abundance”. When you select the latter, a window like the one below opens, and you can set any isotopic composition.
C1.3. Reference Tab
Display/set the information of references that are the source of the crystal structure data.
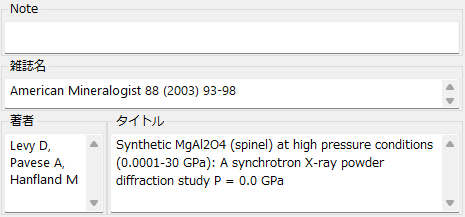
Note
Display/set memos and notes.
Author
Display/set the author names of the referenced paper.
Journal
Display/set the journal name of the referenced paper.
Title
Display/set the title of the referenced paper.
C1.4. Equation of State Tab
Calculate pressure following the equation of state (EOS). For detailed expressions of each EOS model, please refer to the separate page.
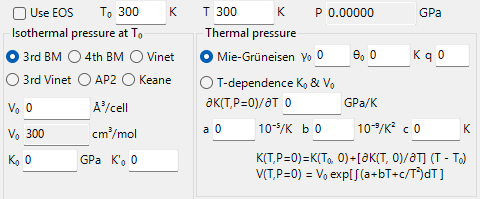
Use EOS
When checked, pressure P is calculated using the entered equation of state.
T0
Set the reference temperature in the equation of state.
T
Set the temperature at the time of crystal volume measurement.
P
Calculate the pressure according to the set parameters and display it in GPa units.
Isothermal Pressure
V0, K0, K’0
Set the unit cell volume (V0), bulk modulus (K0), and first derivative of the bulk modulus (K’0) when the pressure is 0 GPa and the temperature is T0.
3rd/4th Birch Murnaghan, Vinet/3rd Vinet, AP2, Keane
Calculate pressure with the checked model. For detailed expressions of each model, please refer to the separate page.
Thermal Pressure
Mie-Gruneisen
When checked, thermal pressure is calculated using the Mie-Gruneisen equation. Enter the parameters \(\gamma_0, \theta_0, q \).
T-dependence K0 & V0
When checked, thermal pressure is calculated using the Birch Murnaghan equation with temperature dependence.
C1.5. Right-Click Menu

When you right-click, a menu like this appears.
Scattering Factor
Opens a window displaying a list of crystal planes and structure factors. For details, see C.3 Scattering Factor.
Symmetry Details
Opens a window displaying information about symmetry. For details, see C.2 Symmetry Details.
Read from CIF or AMC File
Supports CIF format and AMC format, which is the format adopted by the American Mineralogical Society.
Output as CIF File
Save the current crystal in CIF format.
Restore Lattice Constants to Those at Application Launch
Restore the crystal’s lattice constants to the values when the software first loaded them. Use this when you have accidentally changed the lattice constants in PDIndexer or similar software.
Convert to P1
Convert the space group to P1.
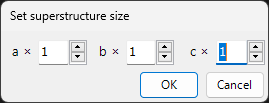
Convert to Superstructure
A window like the following is displayed, and the space group is kept the same while the lattice constants a, b, c are converted to a superstructure by integer multiples.
Convert to Space Group with Different Axes/Origin Setting
Change the space group of the current crystal to a different axis setting or origin choice. A window like the following will be displayed, so select the space group you want to convert to.